Ask for a reprint
email :
* Give your email
2013
ACL
|
C.Martineau, C.Legein, M.Body, O.Péron, B.Boulard, F.Fayon, 'Structural investigation of a-LaZr2F11 by coupling X-ray powder diffraction, 19F solid state NMR and DFT calculations', J. Solid State Chem. 199 326-333 (2013) doi:10.1016/j.jssc.2012.12.016
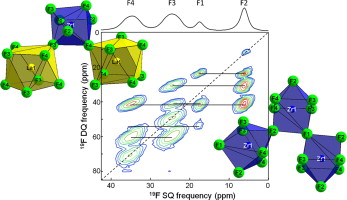
a-LaZr2F11 has been synthesized by solid state reaction. Its crystal structure has been refined from X-ray powder diffraction data (space group n° 72 Ibam, a = 7.785(1) Å, b = 10.086(1) Å and c = 11.102(1) Å). a-LaZr2F11 contains one La, one Zr and four F inequivalent crystallographic sites. F3 and F4 are shared between one ZrF73- polyhedron and one LaF85- polyhedron, while F1 and F2 bridge two ZrF73- polyhedra. 19F 1D MAS NMR spectra of a-LaZr2F11 are in agreement with the proposed structural model. Assignment of the 19F resonances to the corresponding crystallographic sites has been performed on the basis of both their relative intensities and their correlation patterns in a 19F 2D dipolar-based double-quantum recoupling MAS NMR spectrum. DFT calculations of the 19F chemical shielding tensors have been performed using the GIPAW method implemented in the NMR-CASTEP code, for the experimental structure and two PBE-DFT geometry optimized structures of a-LaZr2F11 (atomic position optimization and full geometry optimization with rescaling of the unit cell volume to the experimental value). Computations were done with and without using a modified La pseudopotential allowing the treatment of the 4f localized empty orbitals of La3+. A relatively nice agreement between the experimental 19F isotropic and anisotropic chemical shifts and the values calculated for the proposed structural model is obtained.
|
|