Ask for a reprint
email :
* Give your email
2013
ACL
|
T.Ohkubo, E.Tsuchida, M.Gobet, V.Sarou-Kanian, C.Bessada, Y.Iwadate, 'First-principles molecular dynamics simulation and conductivity measurements of a molten xLi2O–(1-x)B2O3 system', J. Phys. Chem. B 117 5668–5674 (2013) doi:10.1021/jp312486m
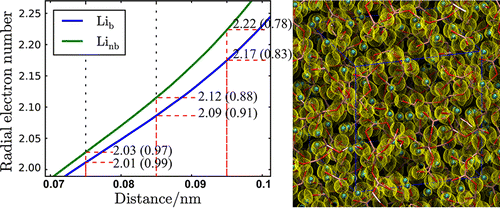
The electronic properties and atomic structure of a molten xLi2O–(1 – x)B2O3 system were investigated by measuring conductivity and using first-principles molecular dynamics (MD) simulations. The conductivities obtained were converted to a Li self-diffusion coefficient Dσ, using the Nernst–Einstein equation to assess charge transfer mechanisms. Dσ was compared with a Li self-diffusion coefficient, DNMR, which we measured in a previous study using high-temperature pulsed field gradient NMR. The DNMR/Dσ of xLi2O–(1 – x)B2O3 (0.2 ≤ x ≤ 0.5) at 1250 K ranged from 2.5 to 3.2, following the same trend as room temperature ionic liquids. First-principles MD simulations were performed using our own finite element density functional theory code, FEMTECK (finite element method-based total energy calculation) for molten xLi2O–(1 – x)B2O3 systems at 1250 K. We found that the O–B–O angle distribution functions were characterized by a peak at approximately 120°. Although the electron number from the electronic radial distribution function was arbitrary with regard to the cutoff distance, the net Li charge calculated from the integrated electron number surrounding Li was approximately 0.9 at 0.085 nm. The mean square displacement (MSD) of Li as a function of time was evaluated from the atomic configuration. Li self-diffusion coefficients calculated from the MSD were in better agreement with experimental results than they were using classical MD.
|
|